Sickle Cell Disease
Key points
- Early recognition of sickle cell disease is key to preventing complications. All children and youth new to Canada who travel from endemic regions and do not have reliable, previous documentation should be screened for sickle cell disease using high-performance liquid chromatography (HPLC), or hemoglobin analysis (e.g., Hb electrophoresis).1
- Children with sickle cell disease require specialized care. Management includes preventing infection, folic acid supplementation due to chronic hemolysis and consideration of hydroxyurea treatment. Referral to a specialized clinic, where available, is recommended.
Epidemiology
The map below shows the global distribution of the frequency of the HbS allele for sickle cell anemia.
Figure 1: Global distribution of frequency of HbS allele for sickle cell disease
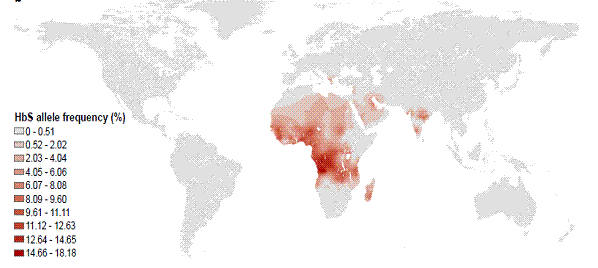
Source: Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. The Lancet 2013;381(9861):142-51. With permission.
Genetics and pathophysiology
Sickle cell disease is an autosomal recessive disease. It is caused by mutation of the β Hb gene, which results in production of an abnormal beta chain of hemoglobin, allowing polymerization of hemoglobin molecules and alterations in red blood cells that take on a characteristic ‘sickle’-shape. These abnormally shaped cells can accumulate in blood vessels and may cause venous stasis and tissue infarction. Sickle cell disease can affect all bodily organs.1
Types and clinical presentation
There are several types of sickle cell disease with very different presentations. Symptomatic sickle cell disease occurs in people who are homozygous (two copies of the mutation, called SS disease) or compound heterozygous (one copy of the sickle gene and another gene resulting in a defect of the beta hemoglobin, e.g., in sickle-beta thalassemia). Individuals who have only one sickle cell gene mutation are referred to as having the sickle cell trait. Having the trait does not result in symptoms. When two people with the trait have a child, there is a 25% chance that the child will have hemoglobin SS sickle cell disease.
The initial presentation of sickle cell disease often occurs following the normal decline of the fetal hemoglobin, usually after the first 6 months of life. Dactylitis (painful swelling of the fingers and/or toes) is often the first presentation.
Complications of sickle cell disease
People with sickle cell disease can experience painful vaso-occlusive crises. Anemia may be exacerbated by aplastic crisis (in the context of parvovirus B19 infection), sequestration crisis (with acute splenic enlargement), or hemolytic crisis. Other complications can include acute pulmonary crisis, pneumococcal sepsis or meningitis, ischemic stroke and gallstones, but all organs can be affected.
Early recognition of sickle cell disease is key to preventing complications. All children and youth new to Canada who travel from endemic regions and do not have reliable, previous documentation should be screened for sickle cell disease using HPLC or Hb electrophoresis.1
Co-existing hemoglobin variants
Other hereditary anemias may coexist with sickle cell anemia, such as hemoglobin C (Hb C) disease in the African population. When Hb C disease coexists with Hb S disease (referred to as SC disease), this can result in similar disease severity to HbSS sickle cell disease. Similarly, hemoglobin E disease is found in Indochina (Cambodia, Vietnam, Laos) and Thailand, and may be associated with microcytic anemia, but is often mistaken for iron-deficiency anemia. Both can be diagnosed by HPLC or Hb electrophoresis.
Treatment
Children with sickle cell disease require specialized care. Management includes special consideration of preventing infection with antibiotics, such as penicillin V, and additional vaccinations, folic acid supplementation due to chronic hemolysis, and consideration of hydroxyurea treatment. Referral to a specialized clinic, where available, is recommended.
Selected resources
- American Academy of Pediatrics, Section on Hematology/Oncology and Committee on Genetics. Health supervision for children with sickle cell disease. Pediatrics 2002;109(3):526-35.
- The Hospital for Sick Children. About Kids’ Health [Handouts]. See Sickle cell disease: A practical guide for parents, and Sickle cell disease: A practical guide for teachers.
- The Hospital for Sick Children. 2006-07 (rev, 2010). Clinical practice guideline. Acute chest syndrome or pneumonia: Guidelines for management in children with sickle cell disease.
References
- American Academy of Pediatrics, Section on Hematology/Oncology and Committee on Genetics. Health supervision for children with sickle cell disease. Pediatrics 2002;109(3):526-35.
Other works consulted
- Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun 2010;1:104.
Reviewer(s)
- Andrea Hunter, MD
- Anna Banerji, MD